Dysregulated metabolism underpins Zika-virus-infection-associated impairment in fetal development
What governs Zika virus pathogenicity? A story of how a mutation in the M protein of ZIka virus led us to understand the molecular underpinnings behind Zika pathogenesis
TRANSCRIPTOMICSPATHOGENESISZIKA VIRUSCELLULAR BIOENERGETICSMETABOLOMICS

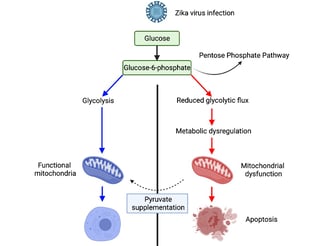
Summary of findings of how a single nucleotide substitution in the Zika virus genome led us to uncover the underlying basis behind Zika pathogenesis. Article by Yau and Low et al., Cell Reports, 2021
Zika has caused a severe pandemic in Brazil, with more than 1,600 babies born with microcephaly from September 2015 through April 2016. It remains a mystery how the Zika virus suddenly became so pathogenic and confined mostly within Brazil.
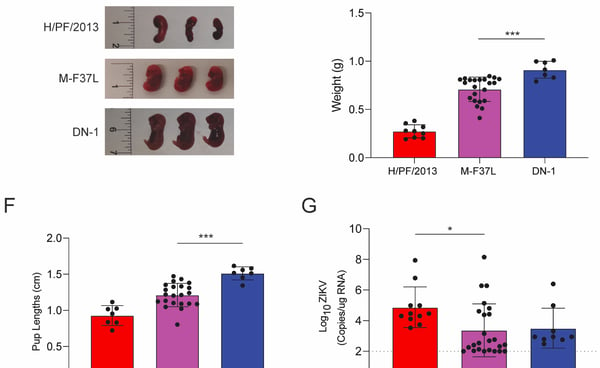
We recently chanced upon a single nucleotide mutation that can increase Zika virulence in vivo. A single mutation in the Membrane protein from phenylalanine to leucine at position 37 (M-F37L) converted a partially attenuated Zika strain (DN1) to one that is pathogenic in interferon knockout mice (A129 mice) and the congenital mouse model (See Figure 1D-1G on the left). Interestingly, the differences in pathogenicity is not attributed to differences in viremia, which led us to investigate the host responses induced by M-F37L and DN-1.


Firstly, a microarray analysis was performed on a primary cell strain (MRC5) infected with different Zika strains, including wild-type (Zika virus strains from Asian and African lineages), M-F37L and attenuated strains to compare the transcriptomic responses between these virus strains. From the principal component analysis (PCA), the M-F37L and pathogenic wild-type Zika virus strains are clustered differently from the uninfected controls and the attenuated strains. Of note, all pathogenic viral strains induced pathways related to inflammation, apoptosis, mitochondrial changes and cell cycle (See Figure 2A-C). Further examination into the gene signatures by Gene-set Enrichment Analysis (GSEA) reveal that the mitochondrial-related pathways were most differentially modulated between M-F37L and DN1. These findings prompted us to characterise the mitochondria of the different Zika strains. Since mitochondria is central in cell metabolism, we also characterised the metabolic profile of cells infected with the different Zika virus strains.




A lot of effort has been placed optimizing the Seahorse Extracellular Flux assay, which allowed us to understand the effects on mitochondria numbers and function after Zika virus infection. This protocol, now published in the STAR methods, produces consistent results between experimental replicates, allowing us to confidently ascertain mitochondria phenotypes after virus infection. We believe this assay could set the foundation for future investigators to use this method to evaluate mitochondria phenotypes within virus infected cells. The mitochondria stress test and glycolytic stress test revealed that the mitochondria oxidative phosphorylation and glycolysis were compromised in the pathogenic Zika virus strains (See Figure 3A, 3C). Furthermore, in collaboration with our colleagues from SMART-MIT, we showed by mass spectrometry that the host metabolism was shunted into the pentose phosphate pathway in M-F37L, resulting in fewer substrates available for mitochondria metabolism, thereby triggering more reactive oxygen species production and inflammation.


These exciting findings led us to postulate if we can have a therapeutic intervention that can rescue mitochondria dysfunction caused by pathogenic Zika virus infection. We used pyruvate supplementation in order to bypass the pentose phosphate pathway and feed directly into the citric acid cycle. Interestingly, mitochondria metabolism was restored, and the transcriptomics signatures related to inflammation and apoptosis were suppressed with pyruvate pre-treatment. Furthermore, ethyl pyruvate pre-treatment partially restored the pathogenic effects caused by wild-type Zika virus strains in A129 pubs (See Figure 5C-G).
Overall, these findings highlight an important aspect of Zika pathogenesis, which involved metabolic derangement that compromises mitochondria functions, consequently increasing inflammation and cell death.
Many open questions remain. Do these results reveal the possibility of using nutrition therapy to relieve the burden of Zika? Are genetic mutations within the Membrane region responsible for increased Zika virus virulence? How do pathogenic Zika virus strains shunt host resources into the pentose phosphate pathway to promote viral replication? We believe that a better understanding of these questions could help us be more prepared of the next Zika virus pandemic if it ever re-appears.
This work is done in Duke-NUS Medical School, and published in Cell Reports. Special thanks to Clement Yau, John Low and Eng Eong Ooi for their significant contribution.